Develop a new method that accurately describes the electron-electron interaction in materials
We develop a new theoretical method that can accurately describe the electron-electron interaction in materials. A rich variety of materials properties originates from the complicated interactions among electrons and nucleus. Theoretical methods to solve their governing equation are called first-principles calculation. We develop a new first-principles calculation method on the basis of the wave-function theory, for accurate description of the complicated and non-trivial materials properties.
FEATURE
We challenge an essential problem in materials science.
One of our main targets is a strongly correlated system with non-trivial electronic structure.
We develop our original calculation method and computational code.
We consider that our method can be an important and fundamental technology for materials science in future.
RESULTS
Research progress
Improvement of the accuracy of our calculation method
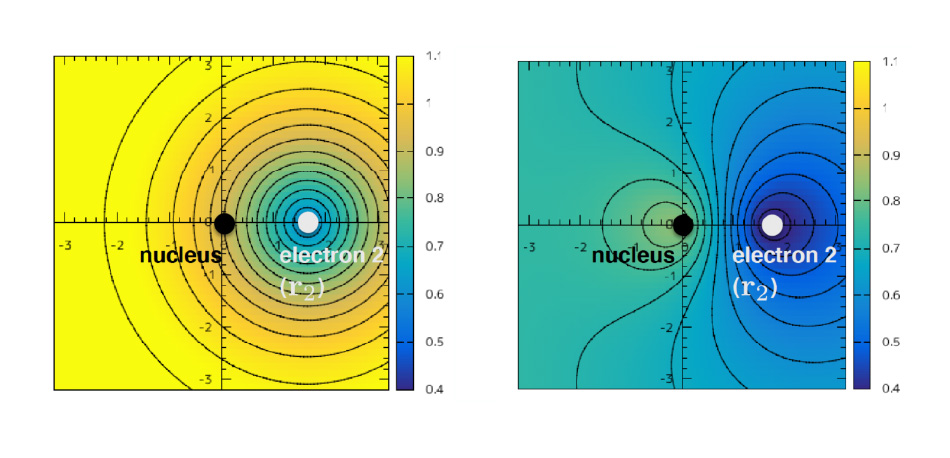
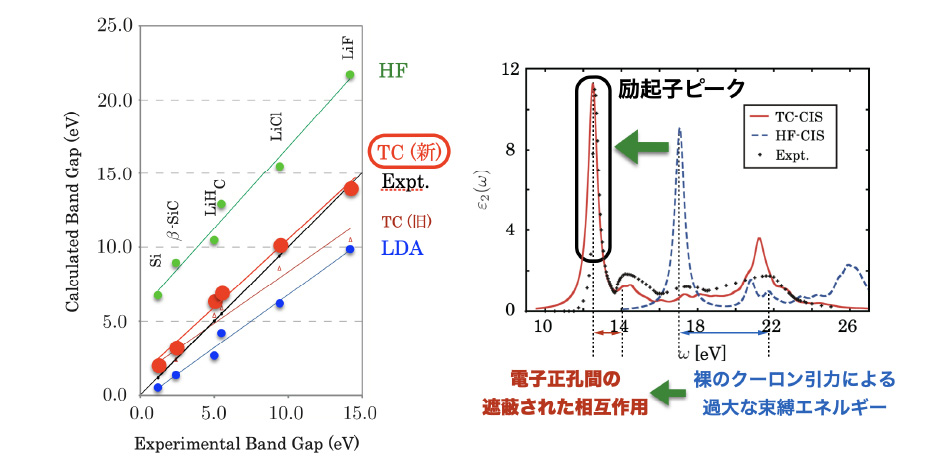
We have developed a new efficient algorithm for our method, improved the accurate correlation factors that describe the electron correlation effects, and obtained accurate optical absorption spectra of solids. While computational cost of the wave-function theories is generally expensive, we have succeeded the efficient computation of our wave-function theory, which has broadened a range of application of our method. We have clarified which feature of the correlation factors improves an accuracy of the electronic-structure calculation. For electronic excited state calculation, we have found that the exciton can be well described by our method, by which we verified a wide applicability of our method.
Further development
Establish a fundamental theoretical method for future materials science
First-principles calculation plays an important role in materials science, both for fundamental science that aims to clarify the microscopic mechanism of non-trivial phenomena, and for applied science that aims to find a high-functional materials. However, the present first-principles calculation methods have several problems such as insufficient accuracy. Our method also has some problems: how to construct the highly accurate correlation factor, how to optimize it, etc. We are trying to resolve these problems and establish a fundamental theoretical method for future materials science.